
Research
It is well established that chromosome instability is an underlying cause of several age-related disorders, including cancer and neurodegenerative diseases. Changes in chromosome number and structure can be triggered not only by DNA damaging agents, but also by intrinsically unstable regions of the human genome itself – which we term the ‘enemies within’. The overarching goal of the Center for Chromosome Stability (CCS) is to functionally annotate these ‘enemies within’, and determine how they drive tissue aging and limit cell immortality.
The CCS currently comprises 7 core research groups. In addition, there are two affiliated groups headed by Andres Lopez-Contreras and Andrew Blackford. Andres was a PI in the CCS until 2020, but now runs a group at CABIMER in Seville, Spain. Andrew was recruited in 2023 from the University of Oxford. The different research groups possess complementary expertise in genetics, biochemistry, structural biology and molecular/cell biology. The leaders of the research groups are listed below, with a short description of research interest and a link to the home page of the group.
"Mechanism of chromosomal fragile site instability"
Our research is focused on understanding how chromosomal instability impacts on human disease. We study genes in human cells that, when mutated, lead to defective genome maintenance and high rates of cancer, neurodegeneration or infertility.
In recent years we have focused on common fragile sites as a model for understanding how ‘difficult-to-replicate’ regions of the human genome affect propagation of the genome through successive cell divisions. As part of these studies, we identified ultra-fine anaphase bridges (UFBs) which are DNA thread-like structures connecting the separating sister chromatids in the anaphase of mitosis. These UFBs exist in every human cell division but are normally ‘invisible’ due to their lack of staining with DNA dyes. They are coated with specific proteins, including the SNF2 family protein, PICH and the BLM helicase (which when mutated causes Bloom’s syndrome – a cancer predisposition disorder), that allows detection using specific antibodies.
We combine protein biochemistry with molecular/cell biology and high resolution imaging of human cells. Latterly, we have also been developing tools to exploit new developments in microfluidics and single molecule biophysical techniques to reconstitute in vitro defined steps in DNA metabolism.
Homepage for Hickson Group
(Department of Cellular and Molecular Medicine)
"Chromosome dynamics in the germline"
Chromosomes are rearranged and organized into new sets to create diversity as they are passed from parent to offspring through the germline. The genetic changes can be followed in populations, however this represents only a small proportion of the diversity that is generated in our germline. Men produce 500 billion sperm in their lifetime and women are born with two million eggs. Human reproduction is particularly error-prone. 50% of pre-implantation embryos have defects in their development and 20%-85% of human eggs have extra or missing chromosomes (aneuploidy). Maternal age is the strongest risk factor known for aneuploidy and our interests in reproductive aging also includes understanding the decreased quality as well as the decline in the number of eggs as women age.
Our laboratory investigates the role of the DNA damage response and cell cycle proteins in governing the genetic changes that occur in the germline to generate diversity and maintain genome stability. In particular, we focus on those genes that when defective give to reproductive disease or cancer. We use a powerful combination of model organisms (mouse and yeast) as well as human eggs and embryos to explore this poorly understood area of human biology.
Homepage for Hoffmann Group
(Department of Cellular and Molecular Medicine)
Our research is multidisciplinary and collaborative, and centers on computational studies of functional, structural, comparative genomics and transcriptomics. We do this to understand
1) the epigenomic changes just before and after mammalian fertilization as well as the effects on embryo fitness, and
2) DNA template conflicts e.g. the replication stress occurring in the first cell division of mammalian embryos.
At conception, we all inherit a fortune. Six billion base-pairs worth of instructions for how to produce each and every specific component and emerging properties of our cells, body-plan, physiology, mind, and more.
These legacy instructions are the software needed to program the matter we consist of, and they are literally ancient. However, while much of our programming was optimized already a billion years ago, other components has only recently been introduced into our genomes.
The quest to adapt to changing environments and conquer ecological niches demands that all organisms include a certain level of creative destruction, so that fitting changes are inherited in the offspring through trial-and-error.
As with any large and comprehensive set of instructions, our genome therefore contains conflicting, ambiguous, repurposed, obsolete, inflated, suboptimal, and even selfish components.
Every genome therefore has to contain and uphold a range of conflicts of interests and trade-offs, and much of the collective action of DNA-bound factors and epigenetic modifications conceivably serve to ensure a coordinated and appropriate use of the genome.
Our overarching passion is to seek understanding of how inbuilt genomic conflicts of interests are handled by mammalian organisms.
A key interest is to understand the remarkable environment existing when the mammalian oocyte is fertilized and become an embryo. During this process, extensive genomic reprogramming happens, epigenetic marks are rewritten on a global scale, and retrotransposons are allowed to be strikingly active and roam more freely than at any other stage of mammalian life – all factors that can threaten the integrity of the genome (Bhowmick, Lerdrup, Mol Cell, 2022; Sankar, Lerdrup, Manaf et al, Nat Cell Biol, 2020; Dahl et al, Nature, 2016, Manaf, Lerdrup et al, submitted).
Our work has also led to development of a comprehensive software suite for data analysis to increase productivity and deeper understanding of genome-wide data (Lerdrup et al, Nat Struct Mol Biol, 2016, http://easeq.net).
"High throughput screening for modulators of genome stability"
In response to DNA damage, the DNA repair machinery is assembled at the site of damage in a highly choreographed manner depending on the chromosomal context, the type of damage, cell cycle phase and other factors.
We study the spatiotemporal organisation of DNA repair processes in the cell nucleus with special focus on homologous recombination (Symington et al. 2014), which plays a key role in the repair of DNA double-strand breaks, restart of stalled or collapsed replication forks, and telomere length homeostasis in telomerase negative cells (Silva et al. 2016). To identify the fundamental biological mechanisms underlying DNA repair, we combine genome-wide cell biological and genetic analyses in the yeast Saccharomyces cerevisiae with more focused studies in vertebrate cell lines (Gallina et al. 2015). In addition to studying the proteins that catalyse DNA repair, we also aim to identify and understand the post-translational modifications that regulate these processes such as sumoylation, phosphorylation, acetylation and ubiquitylation.
If chromosome aberrations are not repaired in a timely manner prior to cell division, they may lead to DNA anaphase bridges in mitosis (see illustration below), chromosome breakage, and ultimately missegregation of genetic material (Germann et al. 2014; Pedersen et al. 2015). DNA anaphase bridges have been linked to chromosomal fragile sites (see the Hickson Lab website), which are chromosome regions prone to exhibit gaps and breaks on metaphase chromosomes, especially when cells are challenged by DNA replication stress. Notably, more than 50% of recurrent cancer mutations have been linked to fragile sites. Our aim is to identify the proteins and mechanisms responsible for sensing and resolving DNA anaphase bridges in a manner that preserves chromosome integrity and to understand the coordination of these processes with mitosis.
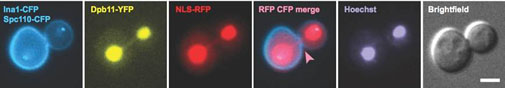
Caption: Dpb11-coated anaphase bridges activate the NoCut checkpoint to delay cytokinesis (Germann et al, 2014).
Homepage for Michael Lisby
(Copenhagen Biocenter)
Our group aims to understand the processes cells use to counteract replication stress caused by either internal factors or external factors, and why some regions in the genome are particularly susceptible to those factors.
1) The analysis of mechanisms underlying MiDAS
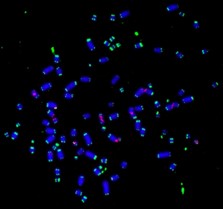
It is well established that incomplete replication can cause a delay in chromatin condensation that leads to the ‘expression’ of common fragile sites (CFSs) [1, 2]. CFSs are hot spots for deletions and chromosome rearrangements in cancer [3]. We previously have taken part in the discovery of a process called mitotic DNA synthesis (MiDAS) that operates in mitosis is a strategy used by human cells to rescue the incomplete replication at those loci, particularly in cancers cells that has elevated replication stress (RS) [4-7] (Figure 1). Particularly, both RAD52 and POLD3 play a crucial role in MiDAS. Our recent findings demonstrate that: i) RTEL1, a DNA helicase, can prevent the accumulation of G-quadruplex-associated R-loops at difficult-to-replicate loci including CFSs in the human genome in S phase, and can facilitate MiDAS in M phase [8]; ii) both translesion polymerases and replication replication polymerase delta play a crucial role in completing MiDAS [9]. In addition, using a BioID strategy [10], we have identified a panel of factors that could potentially work closely with POLD3 when cells are challenged with RS. We are now investigating the functions of these factors and their relevance to MiDAS.
2) The analysis of folate deficiency induced genome instability
Folate deficiency is known to be associated with a diverse range of human disorders including fetal neural tube defects, age-associated dementia, infertility, and some type of cancers. Intriguingly, folate deficiency is known to cause the expression of group of rare fragile sites, all of which contain long stretch of CGG simple repeats. The most well studied locus of this kind is called FRAXA that is associated with Fragile X syndrome (FXS). Using FXS cells as a model, we demonstrated that folate deprivation triggers the extensive missegregation and aneuploidy of chromosome X [11], and MiDAS at the FRAXA locus via the break-induced DNA replication (BIR) that requires the SLX1/SLX4 endonuclease complex, the RAD51 recombinase and POLD3 [12]. Recently, based on a combination of bioinformatic and cellular biology analysis, we demonstrated that folate deficiency could cause the abnormal segregation of a region with CG-Rich trinucleotide repeats on human chromosome 2 [13]. We are currently investigating other regions that are vulnerable to folate deficiency in the human genome, and the strategies cells employ to maintain the stability of those regions (Figure 2).
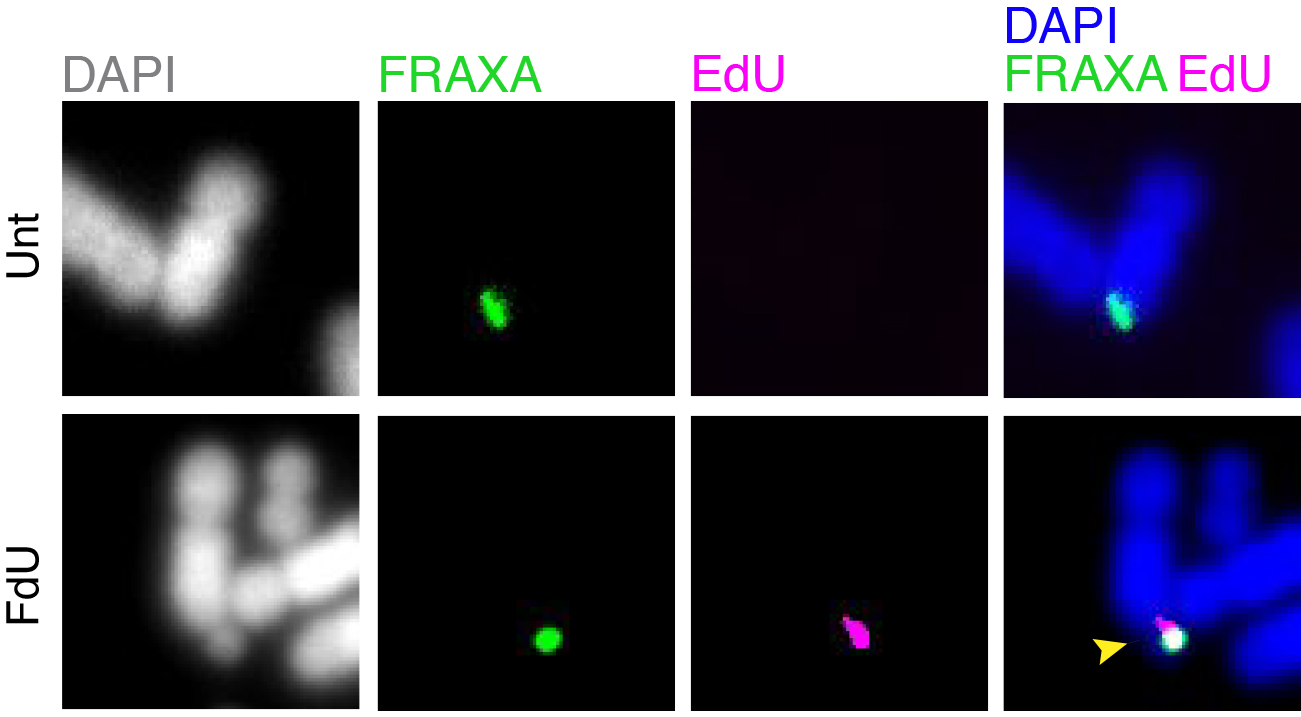
Figure 2. MiDAS occurs at FRAXA in response to folate stress. GM09237 cells, which have over 900 CGG repeats at FRAXA, were treated with fluorodeoxyuridine (FdU) for 17 hours, subjected to EdU incorporation in M phase, and then harvested for metaphase chromosome analysis. The location of FRAXA was validated by a FISH probe. Representative images of FRAXA loci (green) and an EdU locus (magenta) are shown. Yellow arrow indicates a fragile FRAXA locus. Unt, untreated. FdU, cells treated with FdU.
Homepage for Liu Group
(Department of Cellular and Molecular Medicine)
"DNA damage and replication stress signaling"
A key focus of the Mailand lab (The Novo Nordisk Foundation Center for Protein Research) is to understand how regulatory signaling processes promote cellular responses to DNA damage and replication stress to protect chromosome stability in mammalian cells. While much is known about the basic machinery underlying DNA repair pathways, the regulatory framework that controls and coordinates these processes is less well understood. To remedy this knowledge gap, we are combining innovative systems-wide screening approaches with focused cell- and biochemistry-based studies in order to identify and functionally characterize new factors and signaling processes that protect the integrity of the genome following genotoxic insults.
Within the Center for Chromosome Stability (CCS), our major focus is to characterize the signaling processes that enable cells to overcome replication stress, a major driver of genome instability. To understand the molecular mechanisms employed by cells to mitigate the threat posed by replication stress, we are employing cutting-edge strategies to enrich and map the factors that are recruited to the replication machinery when it encounters DNA lesions or other impediments to its continued progression. In one approach, we are using the BioID method for biotinylation and isolation of factors acting directly in the context of replication forks under various conditions. In another approach, we combine Xenopus egg extracts, a powerful cell-free system for biochemical studies of DNA transactions, with mass spectrometry to identify the spectrum of cellular proteins undergoing enrichment at the replication machinery upon DNA damage or replication stress. Using such strategies we are now able to obtain detailed, systematic insights into the signaling processes that operate directly in the context of stalled replication forks to counteract the deleterious consequences of replication stress. In collaboration with other groups within CCS, we are currently studying the functions and physiological importance of several newly identified components of replication stress-responsive pathways in protecting chromosome stability.
Homepage for Mailand Group
(Novo Nordisk Foundation Center for Protein Research)
"The molecular mechanisms of faithful genome replication"
Our lab aims to understand the molecular mechanisms that maintain eukaryotic genome stability during DNA replication using a combination of genetic, biochemical, cellular, and structural techniques, including cryo-EM.
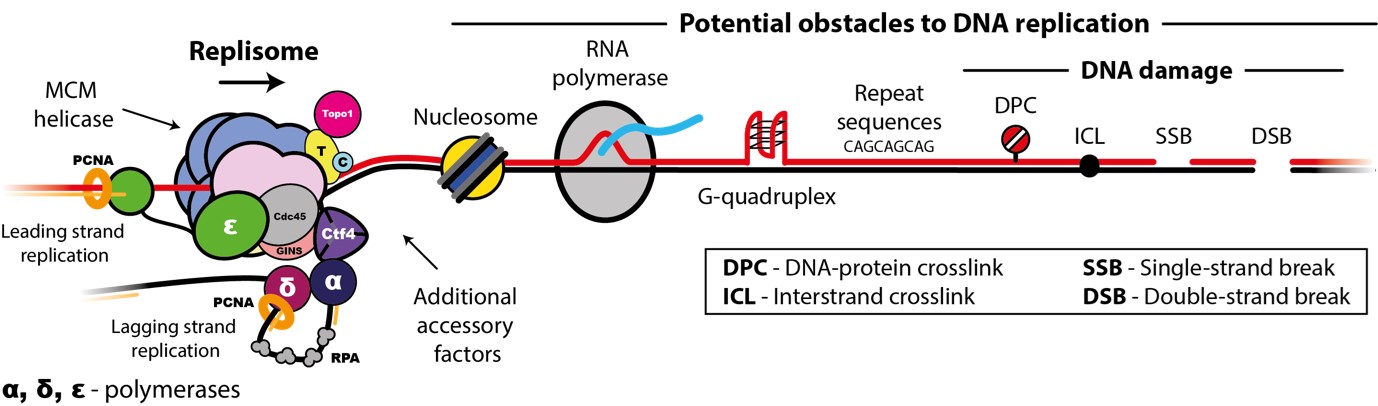
Accurate chromosome replication is essential for controlled cell proliferation and the faithful transfer of genetic information from one generation to the next. A failure to maintain genome stability can cause diverse developmental disorders and age-related diseases, including accelerated neurodegeneration and cancer. Eukaryotic genomes are duplicated by large, multi-component protein machines called replisomes. At their core, replisomes consist of a helicase that unwinds duplex DNA and polymerases that catalyse the synthesis of new DNA molecules according to the parental template. A diverse range of accessory factors are also required facilitate the faithful replication of eukaryotic chromosomes, which contain an array of ‘obstacles’ that could otherwise stall or inhibit the replication machinery. Together, the replisome and accessory factors regulate the timing, speed and accuracy of replication, minimising replication errors and preventing genome instability. Our group aims to understand the molecular mechanisms by which eukaryotic replisomes and accessory factors preserve genome stability during DNA replication and how failures in these mechanisms cause disease. Ultimately, we plan to use the knowledge we gain to develop novel therapeutic approaches to prevent or treat human disorders linked to genome instability.
Homepage for Miller Group
(Department of Cellular and Molecular Medicine)
Affiliated Research Group
"Mouse models of DNA replication stress"
Chromosome instability leads to genetic alterations that can cause cancer and other diseases. To limit these alterations, cells have evolved complex mechanisms that coordinate cellular responses altogether known as the DNA Damage Response (DDR). However, in many cases the DDR is not sufficient to protect our genomes from either exogenous or endogenous insults such as replication-borne DNA damage. The overall goal of our laboratory is to increase our knowledge of the mechanisms that regulate the DDR and the consequences of their deregulation. This will help us to dissect and understand the underlying causes of a number of diseases and to identify novel therapeutic strategies.
With this purpose, we are utilizing a wide range of approaches to investigate chromosomal instability from the underlying molecular mechanisms to its ultimate consequences on health. We are using cellular-based systems to perform screens, gain mechanistic insight and analyse cellular phenotypes in different genetic contexts. In addition, we will generate transgenic mouse models to address the physiological impact of specific genetic alterations and, in particular, their influence on cancer and aging. For both, cellular and mouse models-based studies, we are using the novel CRISPR/Cas9 technology to efficiently manipulate the genome. We also employ high content microscopy (HCM) as a routine approach to quantify alterations in the DDR in our different models. In addition, HCM will allow us to set-up different genetic and drug screenings to identify novel therapeutic targets or compounds with clinical interest. Finally, in collaboration with groups at the Center for Protein Research (CPR), we will perform a number of proteomic studies to investigate novel regulatory networks and the interplay between different proteins involved in the DDR. The most relevant findings will be validated and further characterized using mouse models.
Mutations caused by DNA damage enable a normal cell to become cancerous. This is highlighted by the fact that individuals with mutations in many genes involved in DNA damage recognition, signalling and repair are predisposed to cancer, and that somatically acquired defects in such genes can drive tumour formation. Furthermore, some of the most effective cancer treatments work in tumour cells by inducing DNA damage, particularly DNA double-strand breaks, which are especially toxic and difficult to repair accurately without introducing mutations. Exploiting knowledge of DNA double-strand break repair is therefore likely to lead to more effective and personalised cancer therapies and treatments for patients with DNA repair disorders in future.
The aim of our research is to gain a greater understanding of the signalling mechanisms cells use to coordinate DNA double-strand break recognition and repair with cell cycle checkpoint activation and apoptosis. To achieve this, we are using cutting-edge bioinformatics, proteomics, microscopy and CRISPR-Cas9 gene-editing techniques to answer specific questions related to DNA damage signalling. In doing so, we hope to provide novel insights into carcinogenesis and how it is held at bay by the cell’s DNA damage response system. We also aim to translate our research to develop novel potential cancer treatments. In particular, we are interested in the potential utility of signalling events for use as biomarkers and to identify novel targets in the DNA damage response for anti-cancer drugs.
Homepage for Blackford Group
(Department of Cellular and Molecular Medicine)